Page 4 of 12: Survey of elementary geobiochemistry > the Lithosphere >
The Earth's crust
Formation and evolution of the Earth

The hydrosphere
Page 4 of 12: Survey of elementary geobiochemistry > the Lithosphere >The Earth's crust |
|||||||||
|
Formation and evolution of the Earth |
|
The hydrosphere |
|||||||
The structure and composition of the outer part of the lithosphere has been profoundly affected by interactions with the atmosphere over one-quarter of the surface area of the earth, and with the hydrosphere over the remaining area. Further modification of the outermost parts of the crust has occurred as the result of the activities of living organisms. These changes have transformed much of the outermost parts of the crust into an unconsolidated surface region called the regolith. Further weathering and translocation of soluble substances often results in a sequence of horizons consisting of sediments, soils, or evaporites.
Chemically, the earth’s crust consists of about 80 elements distributed in approximately 2000 compounds or minerals, many of which are of variable composition. Over 99% of the mass of the crustal material is made up of only eight of these elements, however:
|
|||||||||||||||||||||||||
| Table 1: Average amounts of elements in crustal rocks, mg/g. | |||||||||||||||||||||||||
The crust has its origin in the upwelling convection currents that bring mantle material near to the surface at the mid-ocean ridges. The reduced pressure causes it to melt into magma. The magma may solidify before it reaches the surface, forming basalt, or it may energe from the surface in a volcanic eruption. The oceanic crust consists mostly of the simpler silicate minerals, which are said to be basic or mafic. The more evolved, silicon-rich rocks found in the continental crust are known as acidic or sialic.
Oceanic crust is continually being extruded from regions of the plastic mantle that intrude upward to just beneath the ocean’s floor at the mid-ocean ridges. A corresponding amount of this crust is being returned to the lithosphere at subduction zones off the West coasts of the Americas, and in the process pushing up the mountain ranges that lie along these coasts. The subducted oceanic crust is reheated and combined with sedimentary material to undergo partial remelting and reworking; this is believed by some to be the origin of granite. Subduction proceeds at a rate of a few cm per year, and the complete cycle time is on the order of a few hundred million years.
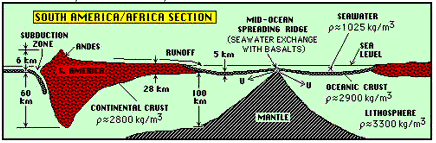 |
|||
|
The continental and oceanic crusts. This schematic cross section runs in a west-to-east direction, from just of the West coast of South America on the left to the African continent on the right. Oceanic crust is shown in black, with with continental crust in red. (From Cosmos and Earth Study Guide, NYU) |
|||
Both oceanic and contintental crusts float on the more dense upper lithosphere, and gradually shift their positions as they push against each other, and in response to the slow convective motions in the medium that supports them. The continental crust is thicker than the oceanic crust, but it is also less dense, which allows it to float higher (and thus to differentiate continents from oceans.) The lower density also prevents it from being subducted. Recycling can occur indirectly as continental material erodes and is deposited as sediments on the ocean floor, but this is a much slower process and one that takes billions instead of millions of years. Some of the very oldest rocks, found in Greenland and Labrador, have been dated at 3.9 billion years, and thus approach the age of the Earth itself.
When magma crystallizes it forms igneous rock, the major component of the Earth’s crust. The crystallization is a complex process which is not entirely understood, due largely to the lack of sufficient thermodynamic data on the various components at high temperatures and pressures. It is known that the different components of magma have differing melting points and densities, and that the phase behavior of multicomponent systems based on some of these substances is quite complex, involving binary and ternary eutectics, solid solutions, the presence of dissolved water (under pressure), and incongruent melting. One consequence of this complexity is that the composition of the magma will change as crystallization takes place; different substances will crystallize at various stages, and the resulting solids may migrate toward the top or bottom of the region if their densities differ greatly from that of the magma
It is well known that larger crystals form when a melt cools more slowly. This principle affords a simple distinction between the coarser-grained plutonic rocks, which are believed to have been formed by gradual cooling of magma pockets within the crust, and the fine-grained volcanic rocks such as basalt. Under the influence of heat and pressure, particularly at plate boundaries, solid crustal material may undergo partial or complete remelting, followed by cooling and transformation into metamorphic rocks such as gneiss, micas, quartzite, and possibly granites.
Granite was once thought to be an igneous rock, originating from the crystallization of a particular kind of magma. The association of granitic rocks with mountainous regions, and the similarity of their compositions in widely scattered regions, lends credence to the more recent hypothesis that granitic rocks are of metamorphic origin.
Another class of rock is sedimentary rock, formed from the consolidation of material produced by weathering and other chemical, and biological processes. Sedimentary rocks cover about three-quarters of the land area of the earth; 80% are shales, 15% sandstones and 5% limestones.
The chemical composition of rocks tends to be complex and variable, and can only be specified in a precise way at the structural level. The traditional way of expressing rock compositions is in terms of the mass percent of the oxides of the elements present in the rock.
|
||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Table 2: Chemical composition of a typical rock The figures are in percent by weight. |
||||||||||||||||||||||||||||||||||||||||||||||||||||
This does not mean that these oxidess, or the structural units they represent, are actually present as such in a rock. In the chemical analysis of rocks, oxygen is generally not determined separately. When it is, however, it is found in an amount that would be expected to combine stoichiometrically with the other elements present. Thus the composition of albite can be written as either NaAlSi3O8 or Na2O·Al2O3·6SiO2 . Some rocks contain varying ratios of certain elements. For example olivine, which can be considered a solid solution of Mg2SiO4 and Fe2SiO4, can be represented by (Mg,Fe)2SiO4; this implies that the ratio of metal to silica is constant, and that magnesium is ordinarily present in greater amount than iron.
The major structural elements of rock (both in the crust and in the mantle) are the silicate minerals, built from silicon atoms surrounded tetrahedrally by four oxygens. The simplest of these consist just of SiO44– tetrahedra interspersed with positive ions to achieve electroneutrality; olivine, (Mg,Fe)2SiO4 is a well known example. More commonly, the silicate groups polymerize by sharing one or more oxygen atoms at adjacent tetrahedral corners. Depending on the number of joined corners per silicate unit, this can lead to the formation of a wide variety of chains (pyroxenes, amphiboles) and sheets (micas), culminating in the complete tetrahedral polymerization that produces quartz, SiO2.
Higher degrees of polymerization are associated with higher ratios of Si to O, smaller quantities of positive ions, and higher melting points. Thus when magma cools, the first silicates to crystallize are the olivines, followed by chain and sheet minerals having progressively higher degrees of polymerization and smaller fractions of cations of metals such as Fe and Mg.
Although some elements are distributed fairly uniformly throughout the crust, others occur at greatly enhanced concentrations in localized areas. There are two general processes that result in these localized excesses, which are called ores when their extraction and refining is economically feasible
The first of these relates to how well a metallic ion can fit into the silicate lattice structure. Ions having the right charge and size can readily enter this structure, displacing the more common ions of Fe, Al and Mg. Such ions (of which Ga3+ is an example) are readily soluble in other minerals and thus are widely disributed and do not concentrate into ores. Other ions may be too large (Cs and La), too small (Li, Be, B) or too highly charged (Nb, Ta, W) to be accommodated in silicate mineral structures; these elements tend to remain in the magma as it solidifies, finally forming solid minerals only in the last stages of cooling.
The other major source of ores is hydrothermal formation. Magma contains some water itself, and additional water from the surface is able to reach the heated rock near magma chambers. At the very high temperatures and pressures that prevail in these regions, the water can dissolve many compounds such as sulfides which are normally considered highly insoluble. When these superheated solutions rise to the surface the solids are re-deposited, often in highly concentrated form. Ores of Cu, Sn, W, and possibly some iron ores, as well as some native metals such as gold, are believed to be formed in this way.
 |
Hydrothermal vents known as “black smokers” have been observed at sites of sea-floor spreading; the “smoke” consists of metallic sulfides which precipitate in the cold seawater. The veins of pyrites (FeS2) and similar sulfide minerals that are often observed in rock formations are the result of hydrothermal solutions that once penetrated cracks and fissures in the rock. The hydrothermal vent photographed here emits both black and white "smoke". |
||||
|
Deep-sea hydrothermal vents (many more pictures) Theories of ore formation (a more technical tutorial) |
|||||
The weathering of rocks at the earth’s surface is a complex process involving both physical and chemical changes. The latter tend in principle to be rather simple kinds of reactions involving dissolution, reaction with carbon dioxide, hydrolysis, hydration, and oxidation. The difficulty in studying them and in arriving at a quantitative description is that these reactions occur very slowly and may never reach an equilibrium state. A comparison of the two rightmost columns in Table 2 on page 14 provides some illustration of the overall effect of these changes, although it must be emphasized that these are relative composition data, and thus cannot show how much of a given component has been lost. In general, sodium, calcium and magnesium seem to be lost more rapidly than potassium and silicon, while iron and aluminum decrease very slowly. Individual rates are of course dependent on the particular structural units containing the element, and also vary somewhat with grain size and condition of the surface.
Water is undoubtedly the most important weathering agent. Not only does it act as a solvent for ionic dissolution products, but it also brings other active agents such as carbon dioxide and oxygen into intimate contact with the rock material. As water percolates into the outermost layers of the crust, it extends the zone of weathering beneath the surface; the effects of this are quite noticeable in a number of buried sedimentary materials such as Paleozoic sandstones, which tend to be depleted of all but the most resistant minerals. Dissolution, the simplest of all the weathering processes, usually results in ionic species, some of which may react with water to yield acidic or alkaline solutions. Dissolution of silica, however, results in the neutral species H4SiO4. Reactions involving hydration and dehydration are very common, and since the free energy changes tend to be small, these reactions can usually take place in either direction under slightly different conditions. Thus gypsum and anhydrite are interconvertable at observable rates under common environmental conditions:
CaSO4·2H2O ![]() CaSO4 + 2 H2O
CaSO4 + 2 H2O
In many cases, however, the reaction products are not very well characterized, thermodynamic data is lacking, and the reactions proceed so slowly that they are not entirely understood. For example, both hydrous and anhydrous iron oxides can be found in similar geologic environments, but the little is known about the interconversion process, represented approximately as
Fe2O3 + H2O ![]() 2 FeOOH
2 FeOOH
Solid carbonates tend to dissolve in acidic solutions, including those produced when atmospheric carbon dioxide dissolves in water. Thus the major surface limestone deposits (largely CaCO3, with some admixture of MgCO3) tend to be highly eroded in non-arid regions, and the local groundwater may have Ca2+ as high as 0.1-0.2M. Thermodynamics can unambiguously predict the most stable oxidation state of a metal ion under given conditions of pH and oxidant concentration. The mechanisms tend to be very uncertain, however. For one thing, both the reactant and product can often exist in various states of hydration, and the dissolved species (which probably undergo the actual oxidation) often consist of polycations and complexed species.
Compounds of Fe(II), for example, will always tend to oxidize to Fe(III) in the presence of air; the various oxides of iron are responsible for the bright colors seen in many geological formations, and in certain soils. Some of the net reactions that probably occur are
Fe2SiO4 + 1/2 O2 + H2O ![]() Fe2O3 + H2SiO4
Fe2O3 + H2SiO4
2 FeCO3 + 1/2 O2 ![]() Fe2O3 + 2 H2CO3
Fe2O3 + 2 H2CO3
An environmental side effect of the first process is the release of hydrated silica. Also, where both starting materials are present, the carbonic acid produced in the second reaction is believed to promote the dissolution of ferrous silicate, creating a source of Fe(II) ions that can be rapidly oxidized:
Fe2SiO4 + 4 H2CO3 ![]() 2 Fe2+ + 4 HCO3– + H4SiO4
2 Fe2+ + 4 HCO3– + H4SiO4
Fe2+ + 4 HCO3– + 1/2 O2 ![]() Fe2O3 + 4 H2CO3
Fe2O3 + 4 H2CO3
The oxidation of sulfides can produce strongly acidic solutions:
2 FeS2 + 15/2 O2 + 4 H2O ![]() Fe2O3 + 2 SO42– + 8 H+
Fe2O3 + 2 SO42– + 8 H+
The effects of this can be seen in formations containing outcrops of pyrite veins, where the surrounding rocks are heavily stained with yellow and brown Fe(III) oxides, and the groundwater tends to be highly acidic. This process is mediated by microorganisms, and is an important source of acid pollution associated with mines and mine tailings.
The various components of rocks weather at different rates. The more basic components such as CaO and MgO tend to disappear first, especially if in contact with groundwaters containing high CO2 concentrations. For rocks in general, the first reaction is usually hydration, followed by hydrolysis which can be summarized by
4 KAlSi3O8(s) + 22 H2O ![]() Al4Si4O10(OH)8(s) + 8 H4SiO4(aq) + 4 K+ + 4 OH–
Al4Si4O10(OH)8(s) + 8 H4SiO4(aq) + 4 K+ + 4 OH–
in which other Group 1 or 2 cations might replace potassium. The product Al4Si4O10(OH)8 is kaolinite, a form of clay (see below).
In general, the rocks which crystallized first from the magma (the Ca-feldspars and olivines) weather more rapidly than do the lower-melting rocks.
 |
|
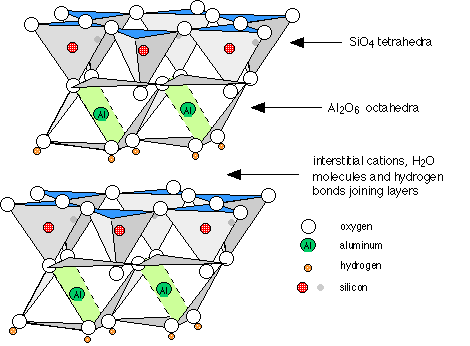
Clays are the solid end products of the weathering of rocks. They are basically composed of alternating sheets of “SiO2” and “AlO6” units in ratios of 1:1 (kaolinite), 2:1 (montmorillonite and vermiculite) and 2:2 (chlorite). In between the sheets, and holding them together by hydrogen bonding are water molecules. Also present are cations such as K+, Ca2+ and Mg2+ which act to neutralize the negative charges of the oxide ions.
 |
|||
|
Structure of a clay, showing two layers of the stacked sheets of kaolinite. |
|||
The major agents of physical weathering of exposed rocks are rapid changes in temperature (promoting fracture by differential expansion), the abrasive action of windborne material and glacier movement, and especially by the penetration of water into cracks and its subsequent freezing. The expansion of water on freezing can exert a pressure of 150 kg cm–2, whereas the tensile strength of a typical rock is around 120 kg cm–2 . The roots of some plants are able to penetrate rock quite effectively, producing comparable expansive pressures in subsurface rocks.
|
Soils are a product of the interaction of water, air, and living organisms with exposed rocks or sediments at the earth’s surface. A typical soil contains about 45% inorganic solids and 5% organic solids by volume. Water and air each make up about 20-30%. A simple way of classifying soils is based on the relative quantities of clay, silt and sand in the solid component. For ordinary agricultural purposes, loams are considered the most ideal soil type. |
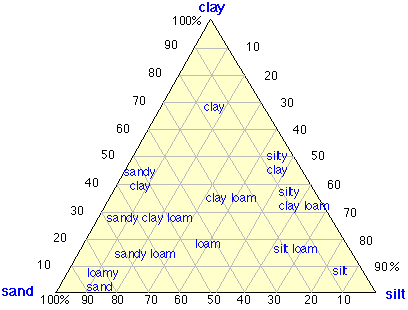 |
||
The primary inorganic components of soils consist of sand and silt particles that come directly from the parent rocks. This fraction is dominated by quartz and feldspars (aluminosilicates). Secondary components are formed by chemical changes within the soil itself, or in sediments from which the soil derived. These are most commonly clays, but may also include calcite, gypsum, and sulfide minerals such as pyrites; the latter are formed by bacterial action under reducing conditions in the presence of organic matter.
The clays have an especially important effect on both the physical properties of the soil, and on its ability to store plant nutrients, including trace nutrients such as Mo and Mn. These properties are due to the high ion-exchange capacity of clays. The more highly charged cations such as Al3+ and Fe3+ tend to be more strongly absorbed within the inter-sheet regions than do Mg2+ or K+. As plants withdraw these latter cations from the soil water, more are released by the clay components, which thus act as nutrient reservoirs.
The ion-exchange properties of clays also help to maintain the pH balance of soils, through the exchange of H+ and cations such as Ca2+. The soil pH, in turn, strongly affects the solubility of nutrient cations, and thus their availability to plants. For example, the uptake of phosphorus (in the form of H2PO42– , is only efficient within the rather narrow pH range between 6 and 7. Below 6, dihydrogen phosphates of Fe and Al are precipitated, while insoluble Ca3(PO4)2 forms at higher pH’s.
Part of the organic matter of soil consists of organisms (mainly bacteria and fungi) and roots and root hairs. The remainder is largely in the form of fulvic and humic acids. These substances of indefinite composition are classified on the basis of their solubility behavior; fulvic acids remain in solution at pH 2, but humic acids, having molecular weights of 20,000 to 100,000, are precipitated. Both are flexible polyelectrolytes that interact strongly with their own kind and with inorganic ions.
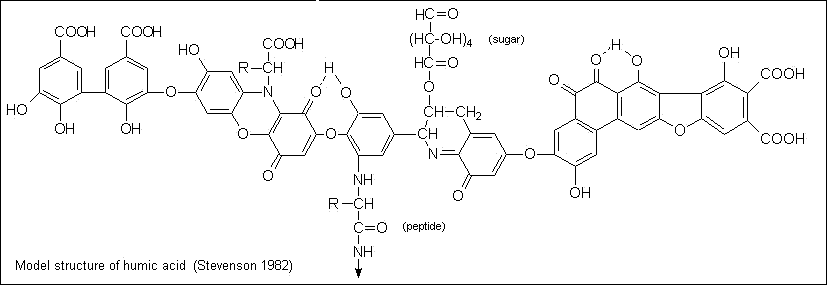 |
||
Associated with the fulvic and humic fractions are a wide variety smaller molecules such as alkanes, amino acids, amino sugars, sulfur and phosphorus derivatives of sugars, etc. Part of the organic carbon in a fertile soil is recycled in 1-2 years; plant residues, which are the major source of soil organic matter, have a half-life of days to months. Once carbon gets incorporated into humic substances, it is locked into a much slower recycling process; the turnover times of fulvic acids are a hundred years or more, while those for fulvic acids are around a thousand years. For this reason, humic substances are the major reservoir of organic carbon in soil. Organic matter, particularly polysaccharides, binds strongly to the cation components of clay colloids; the two together act as cementing agents and strongly influence the consistency and structure of the soil.
Soil water is held by capillary action and adsorption with varying degrees of tenacity. This water binding strength is traditionally expressed in terms of the pressure, or “tension” that would be required to force the water out of the soil. The tension of capillary water varies over a wide range of 0.1-32 atm; only in the lower half of this range will it be available to plants, which can exert an osmotic pressure of up to about 15 atm. Water in excess of the capillary capacity fills larger voids and is called gravitational water. Its presence in surface soils corresponds to a flooded condition that inhibits plant growth by reducing soil aeration.
The gas phase within soil pores generally has a CO2 content of 5-50 times that of the atmosphere due to the action of organisms. O2 tends to be depleted to roughly the extent that CO2 is present in excess. Under conditions of poor aeration (i.e., exchange with the atmosphere), considerable quantities of N2O, NO, H2, CH4, C2H4, and H2S may also be present.
|
What is soil? An introductory survey
Soil science tutorial from U. of Alberta |
|||
Page last modified: 21.01.2008
© 1998 by Stephen Lower
For information about this Web site or to contact the author,
please see theChem1 Virtual Textbook home page.

This work is licensed under a Creative Commons Attribution-NonCommercial 2.5 License.
| O | 466,000 |
| Si | 277,200 |
| Al | 81,300 |
| Fe | 50,000 |
| Ca | 36300 |